

USFDA reduces data requirement for Interchangeability: Significant development for market momentum
Total Number of Biosimilar Approvals in Europe (per Year)
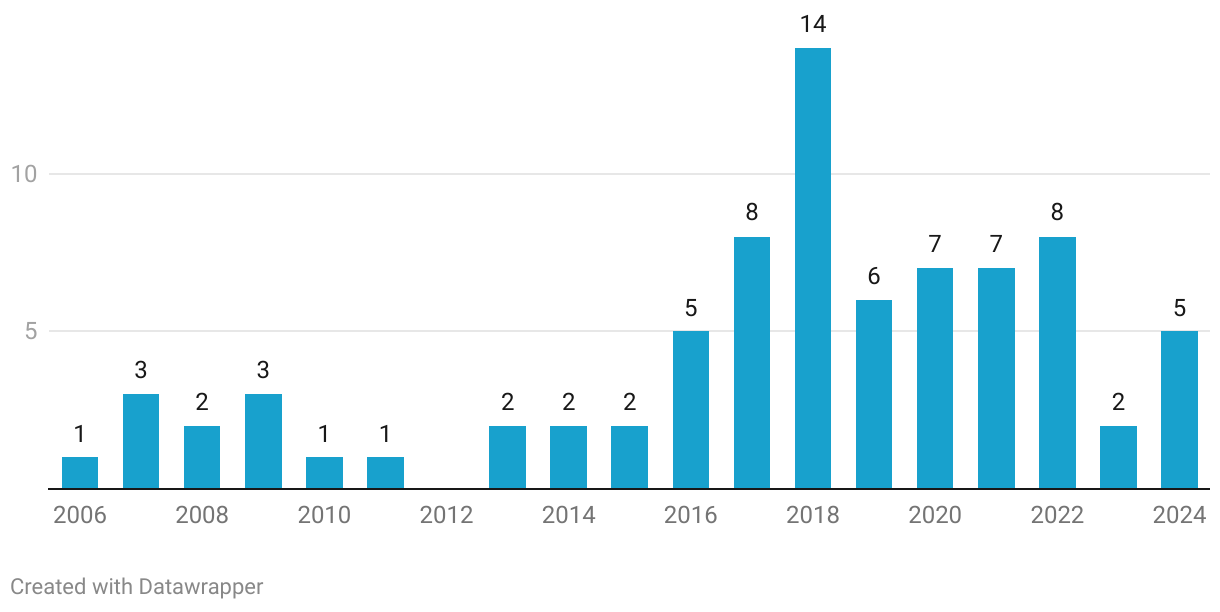
However, of these, only a limited number have been classified as interchangeable by the FDA. Interchangeability has been a sore thorn in the market creation for biosimilars in the US for a long time now. While EMA left it to member states to designate interchangeability, USFDA took on the onus of doing so. This has had significant commercial ramifications with providers more comfortable switching patients to biosimilars when designated interchangeable. In a multi-payor environment, combined momentum from shift from across payors and providers is critical for market creation at large.
In this context, the latest guidance further reducing the burden of evidence needed for achieving the interchangeability designation for biosimilar approvals is likely to have far reaching implications including:
- Lower development costs: By potentially waiving the need for extensive clinical trials, developers can save significant resources.
- Faster approvals: Streamlined processes could lead to quicker biosimilar approvals with the interchangeability designation included, bringing these potentially lower-cost options to patients sooner.
- Increased investment in the biosimilar pipeline: With a more favorable regulatory landscape, biosimilar developers may be more likely to invest in bringing new biosimilars to market.
Biosimilars Landscape: 2024 and Beyond
We anticipate momentum in biosimilar investments to continue unabated. Market potential and pipeline opportunity is high with market opening around the horizon for molecules such as pembrolizumab and nivolumab. The new guidance is likely to further bolster industry confidence for investments in the segment. With a widening base of industry investments, this could potentially lead to:
- Increased competition: More interchangeable biosimilars in the market could drive down costs for patients and payers. At the other end, this will also impact investment business plans and will increase risk of more rapid price erosion that industry will need to be prepared for.
- Improved patient access: Easier access to interchangeable biosimilars could benefit patients who rely on these therapies for chronic conditions. This will be enabled by both factors – higher probability of obtaining interchangeability designation and wider industry investments and more likely biosimilar approvals.
The commercial opportunity around biosimilars is finally more tangible. The long expected impact on patient access is hopefully also a more tangible reality.
Author:

Pushpa leads SMC’s healthcare advisory practice and serves on the Governance Board of the Medicines Patent Pool. SMC has advised several companies to shape their biosimilar portfolios, partner and license biosimilars for global or regional rights and forge commercial partnerships for expanded global reach of their biosimilar investments. Reach us at healthcare@sathguru.com

