

With biosimilar competition intensifying in key markets such as US and Europe, more innovators are strategically deploying formulation and/or delivery innovation for lifecycle management to defend their turf. FDA approval of Roche’s Susvimo® this month reaffirms this trend that we believe is only going to get more intensive in the near to mid-term horizon. Susvimo® represents greater complexity in drug delivery devices and the trend expanding beyond oncology into ophthalmology. We foresee that delivery innovation will be a critical focus for both innovators and biosimilar companies each with their own objective. While guarding brand franchises will motivate innovator investment to this end, aggressive competition amongst biosimilar companies and rapid price erosion are spurring investments from biosimilars companies.
 Susvimo® is a first-of-its-kind therapeutic Port Delivery System (PDS) with ranibizumab 100 mg/ml, first treatment for intravitreal use via ocular implant for neovascular or wet age related macular degeneration (nAMD). It provides an alternative to the current standard method of care – monthly injections for treatment of nAMD, a potentially blinding condition. The port delivery system will deliver the drug to the eye using a refillable implant, with the capacity of continuous release of medicine for period of 6 months. This will reduce number of treatment cycles to 2 per year. The system consists of a refillable reservoir technology, inserted in the eye through pars plana. The titanium release control element of the implant that extends into the vitreous cavity controls ranibizumab diffusion rate from the reservoir. The Phase III trial on nAMD patients, Archway study confirmed non-inferiority vs monthly injections with 98% of trial participants on Susvimo® receiving a refill after 6 months.
Susvimo® is a first-of-its-kind therapeutic Port Delivery System (PDS) with ranibizumab 100 mg/ml, first treatment for intravitreal use via ocular implant for neovascular or wet age related macular degeneration (nAMD). It provides an alternative to the current standard method of care – monthly injections for treatment of nAMD, a potentially blinding condition. The port delivery system will deliver the drug to the eye using a refillable implant, with the capacity of continuous release of medicine for period of 6 months. This will reduce number of treatment cycles to 2 per year. The system consists of a refillable reservoir technology, inserted in the eye through pars plana. The titanium release control element of the implant that extends into the vitreous cavity controls ranibizumab diffusion rate from the reservoir. The Phase III trial on nAMD patients, Archway study confirmed non-inferiority vs monthly injections with 98% of trial participants on Susvimo® receiving a refill after 6 months.
The PDS technology was the brainchild of ForSight Vision4 Inc. Since 2010, Genentech had partnered with the venture for the ocular delivery of ranibizumab and, later in 2017, Roche acquired the company. Susvimo® represents a next gen solution to Roche’s already marketed drug Lucentis (ranibizumab injection), approved to treat nAMD and other retinal diseases by FDA. Roche is also conducting an extension study for Susvimo®, evaluating refill-ability of the device every 9 months or 36 weeks. Along with increasing the duration for refilling the implant, Roche is also exploring other indications including diabetic macular edema (DME) and diabetic retinopathy without DME. European Medicine Agency (EMA) is also currently reviewing the port delivery system for treatment of nAMD. Pricing announced by Roche is comparable to current list price benchmark of Lucentis®: Susvimo® will be priced at $9,250 for the first six months and refills being priced at $8,000, while current list price of vial for monthly injection of Lucentis® is around $1,230.
This development is critical in the light of emerging biosimilar competition. US FDA has already approved the first biosimilar to ranibizumab, developed by Samsung Bioepis. With other biosimilars in the pipeline, price and market share erosion for Lucentis® is difficult to avoid. Susvimo® offering benefit of greater convenience with fewer visits to the hospital is critical for Roche to extend monetization opportunity of this product franchise. Susvimo® will also provide Roche greater competitive advantage against other available treatments for nAMD such as Eylea® (aflibercept) and Beovu® (brolucizumab).
Competition continues to brew in nAMD market and is driving formulation innovation: Competition is stiff in the nAMD space, as Regeneron is fighting back to defend Eylea’s market share with its own high-dose (8mg) aflibercept that requires administration every 3 to 4 months compared to the currently marketed 2mg dose of aflibercept and is indicated for nAMD and macular edema. The high-dose aflibercept formulation is currently under phase 3 development and Regeneron recently announced positive topline phase 2 data which demonstrated the safety, efficacy and tolerability of the 8mg aflibercept formulation against 2mg aflibercept formulation in 106 treatment-naive patients with nAMD.
Until now, Eylea® had dominated the nAMD market in the US with $5 billion in sales in 2020 vs $1.6 billion for Lucentis. Susvimo’s launch could potentially tilt the market share composition by stealing share from the high-dose aflibercept. It could potentially also steal share from off-label bevacizumab market used in nAMD.
Roche has been one of the pioneers in biologic formulation innovation as a strategy for product lifecycle management. For Herceptin® (trastuzumab), it launched two novel formulated versions – Hylecta® in 2019, and Phesgo® in 2020. Hylecta® was a subcutaneously administered trastuzumab formulation containing hyaluronidase that allows quicker dispersion and absorption of large biomolecules in the blood stream – it can be administered in 2 to 5 minutes vs 20 to 90 minutes of intravenous infusion of Herceptin®. The hyaluronidase technology, called Enhanze® was the brain child of Halozyme Therapeutics, which collaborated with Roche since 2006 onwards to develop Hylecta®. Phesgo®, is also based on the same Enhanze® technology and contains pertuzumab in combination with trastuzumab, it’s the first combination monoclonal antibody solution and can be administered subcutaneously in 5 to 8 minutes compared to the regular 150 minutes for a sequential dosing of pertuzumab and trastuzumab separately through intravenous infusion.
With a similar value proposition, Alexion launched an advanced 100mg/ml formulation of Ultomiris® (ravulizumab), with a 60% lesser infusion time than the original 10mg/ml formulation. The advanced formulation was strategically positioned as an improved version of Soliris® (ecuzilumab) in the hope of switching patients from Soliris® and Ultomiris® and wade-off the potential biosimilar competition 2025 onwards.
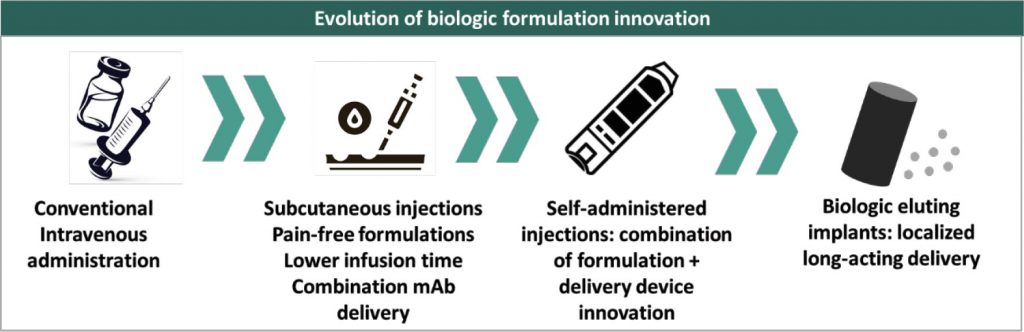 Biologic formulation innovation has until now been pioneered by innovators because of the regulatory complexity in US market to get marketing authorization for a newer formulation of a biosimilar if the same hasn’t been approved for the innovator biologic. Hence, biosimilar developers have often pursued strategy of obtaining approval for the same delivery format as innovator as their first biosimilar and then pursuing delivery innovation as a follow-on supplemental application. This negates the need for a 351(a) biologic application that could imply a substantially higher threshold of clinical data generation as a new biologic. Celltrion is the sole biosimilar developer to adopt a first mover approach for formulation innovation in a biosimilar – it developed the first subcutaneous formulation of infliximab that can be self-administered using a pre-filled syringe or a pen. In the EU market, Celltrion already held a marketing authorization for the intravenous version and later followed that with the registration and launch of subcutaneous formulation. While the EMA approved subcutaneous version as a supplement to the intravenous formulation, the US FDA requires submission of a BLA and the subcutaneous version is currently under clinical development for the US market.
Biologic formulation innovation has until now been pioneered by innovators because of the regulatory complexity in US market to get marketing authorization for a newer formulation of a biosimilar if the same hasn’t been approved for the innovator biologic. Hence, biosimilar developers have often pursued strategy of obtaining approval for the same delivery format as innovator as their first biosimilar and then pursuing delivery innovation as a follow-on supplemental application. This negates the need for a 351(a) biologic application that could imply a substantially higher threshold of clinical data generation as a new biologic. Celltrion is the sole biosimilar developer to adopt a first mover approach for formulation innovation in a biosimilar – it developed the first subcutaneous formulation of infliximab that can be self-administered using a pre-filled syringe or a pen. In the EU market, Celltrion already held a marketing authorization for the intravenous version and later followed that with the registration and launch of subcutaneous formulation. While the EMA approved subcutaneous version as a supplement to the intravenous formulation, the US FDA requires submission of a BLA and the subcutaneous version is currently under clinical development for the US market.
With the level of competition and price erosion emerging as a common reality in biosimilars, formulation innovation is becoming increasingly critical for biosimilar companies. It is emerging as a driver of quest for market share as well as reimbursement support in the current commercial landscape. Enabling partnerships with platform technology companies are thus becoming critical as well. We anticipate more active pursuit of innovation as well as enabling partnerships and expanding thrust from biosimilar companies. More such enabling partnerships are anticipated in the near-to-mid-term.
 Taking another step towards affordable access to biologic drugs, US FDA granted interchangeability designation to Boehringer Ingelheim’s (BI) adalimumab biosimilar (Cyltezo®). The biosimilar was approved by the US FDA in 2017. This was followed by a patent lawsuit against Abbvie which BI ultimately settled for a 2023 launch, like many other biosimilar candidates in the pipeline. However, BI has now scored an interchangeable designation which makes it a formidable contender against other biosimilars set for launch since it can now be automatically substituted at the pharmacy level.
Taking another step towards affordable access to biologic drugs, US FDA granted interchangeability designation to Boehringer Ingelheim’s (BI) adalimumab biosimilar (Cyltezo®). The biosimilar was approved by the US FDA in 2017. This was followed by a patent lawsuit against Abbvie which BI ultimately settled for a 2023 launch, like many other biosimilar candidates in the pipeline. However, BI has now scored an interchangeable designation which makes it a formidable contender against other biosimilars set for launch since it can now be automatically substituted at the pharmacy level.
The interchangeability designation is a landmark achievement for the biosimilars industry since it’s the first monoclonal antibody to have been granted the interchangeability designation. The first biosimilar to be granted interchangeability being insulin analog Semglee®, which we covered in detail in our PharmForward post. The interchangeability designation also grants Cyltezo® one-year of exclusivity, meaning no other interchangeable biosimilar can be launched for up to one year post Cyltezo’s launch.
Humira® was initially launched by Abbvie in a 40mg/ 0.8ml formulation, however, with patent cliff approaching closer, Abbvie launched a high-concentration 40mg/ 0.4ml in a citrate-free formulation which causes lesser pain and requires lower volume administration. The strategy worked in favour of Abbvie since US FDA biosimilarity regulations interpret strength as the concentration of the formulation and a biosimilar must have the same concentration as the reference product, in spite of having the same total drug content (here, 40mg). High-concentration formulation sales increased rapidly in the US, accounting for 66% of US sales in January, 2020. Currently approved biosimilars for adalimumab, including BI’s interchangeable biosimilar are all low-concentration versions and with the market having graduated to the high-concentration version, adoption may be slow in spite of interchangeability.
Both Celltrion and Alvotech are developing a high-concentration adalimumab biosimilars and Alvotech is also aiming for an interchangeability designation in the US market. Alvotech recently announced positive results from a switching study conducted between AVT02, high-concentration adalimumab biosimilar (100mg/ml) and Humira®, which demonstrated no significant differences in clinical efficacy, safety or immunogenicity between the two cohorts. The switching study is to support the regulatory approval as an interchangeable biosimilar.
With regulatory requirements for interchangeability now established and the regulator having more comfort around the same, approval of more interchangeable biosimilars will be facilitated in the near future. This adds on to the series of recent efforts being undertaken by the regulatory agencies and the government to support the development of biosimilars and improve patient access. This adds to the commercial momentum and enhances value realizability on biosimilar investments targeting the US market.
Immune checkpoints play an important role in the immune system, regulating the strength of the immune response in order to prevent destruction of healthy cells. When proteins on the surface of immune cells, T cells, recognize and partner with the proteins on other cells like tumour cells, a signal is sent to the T cells preventing them from destroying the cancer cells. Immune checkpoint inhibitors work by blocking the checkpoint proteins, preventing them from binding with the partner protein, and T cells destroy the cancer cells through the release of new immune responses and/ or enhance existing ones. In 2011, the U.S. Food and Drug Administration (FDA) approved, the CTLA-4-blocking Yervoy® (ipilimumab), the first checkpoint inhibitor immunotherapy for the treatment of cancer—for melanoma and post that seven other checkpoint inhibitors have been approved for diverse indications.
I n October, AstraZeneca released positive results from the TOPAZ-1 Phase III trial, which demonstrated that Imfinzi® (durvalumab), in combination with standard-of-care chemotherapy is both well tolerated and delivers superior clinical benefit compared to chemotherapy alone as a first-line treatment in patients with advanced biliary tract cancer (BTC). BTC is a rare and aggressive form of gastrointestinal cancer that originates in the bile duct or the gall bladder. Imfinzi® is the first immunotherapy to improve the progression-free survival rates in BTC, where the five-year survival rates are as low as 5% to 15%. There had been no advancements in the first-line treatment of BTC for over a decade and this development marks a significant achievement in the treatment of BTC and promises better prognosis for patients.
n October, AstraZeneca released positive results from the TOPAZ-1 Phase III trial, which demonstrated that Imfinzi® (durvalumab), in combination with standard-of-care chemotherapy is both well tolerated and delivers superior clinical benefit compared to chemotherapy alone as a first-line treatment in patients with advanced biliary tract cancer (BTC). BTC is a rare and aggressive form of gastrointestinal cancer that originates in the bile duct or the gall bladder. Imfinzi® is the first immunotherapy to improve the progression-free survival rates in BTC, where the five-year survival rates are as low as 5% to 15%. There had been no advancements in the first-line treatment of BTC for over a decade and this development marks a significant achievement in the treatment of BTC and promises better prognosis for patients.
Roche’s Tecentriq® (atezolizumab) also received a US FDA approval, in October, as an adjuvant treatment for people with early non-small cell lung cancer (NSCLC) making it the first and only cancer immunotherapy approved for NSCLC in the adjuvant setting. Results from its Phase III IMpower010 study showed, treatment with Tecentriq® post-surgery and chemotherapy reduced the risk of disease recurrence or death by 34%.
Immune-checkpoint inhibitors have been a hot area of deal-making and investments in the past year and has seen expanded engagement from big pharma. Earlier this year, GSK signed a potential $2 billion deal with iTeos Therapeutics for the rights to its anti-TIGIT mAb EOS-448, a drug developed for advanced solid tumors, and will undergo clinical trials in combination studies with GSK’s anti-PD-1 therapy Jemperli®. Combination monoclonal antibodies delivering superior clinical outcomes has been a strategy that has been deployed for multiple immune checkpoint inhibitors and potentially expands the clinical use-case. In January, Sanofi struck a potential deal of $1,124 million with Biond Biologics to acquire global rights to anti-ILT2 mAb BND-22. Following this, was another deal between Bristol Myers Squibb (BMS) and Agenus, granting BMS an exclusive global license to, AGEN1777, a bispecific antibody that blocks TIGIT and a second undisclosed target, valued at $1,560 million. The global pipeline of novel immune checkpoint inhibitors is also robust: Immutep is conducting clinical trials for LAG-3 in patients with melanoma, advanced pancreas cancer, metastatic breast carcinoma, non-small cell lung carcinoma and head and neck carcinoma patients. Tesaro, now a subsidiary of GSK is also conducting clinical trials for TSR-022 (Cobolimab), a novel IgG4 anti-TIM-3 mAb, in patients with metastatic NSCLC. Other companies working on novel immune checkpoint inhibitors include Incyte Corporation, Bristol-Myers Squibb, Jiangsu HengRui Medicine, and several universities.

